
在醫療器材產業,ISO 13485《醫療器材品質管理系統標準》曾被視為一張進入國際市場的「門票」。但現在,這張門票的內容雖然沒變,球場的「賽規」卻在快速進化。
目前國際標準趨勢顯示,ISO 13485 在短期內傾向維持穩定,以避免改版引發各國法規體系的連鎖震盪。然而,這對業者來說卻是個挑戰:當 ISO 條文原地踏步,但美國 FDA 的 QMSR、歐盟 MDR 甚至台灣的 QMS 準則都在延伸附加要求時,我們該如何自處?
隱形的力量:當標準不再只是標準
事實上,ISO 13485 已演變為全球醫材法規的核心靈魂。各國不再盲目修訂標準,而是採取「以 ISO 13485 為主體,外掛國別要求」的策略。這意味著,僅僅「符合標準」已不足以應對稽核,您必須看見條文背後的法規地圖:
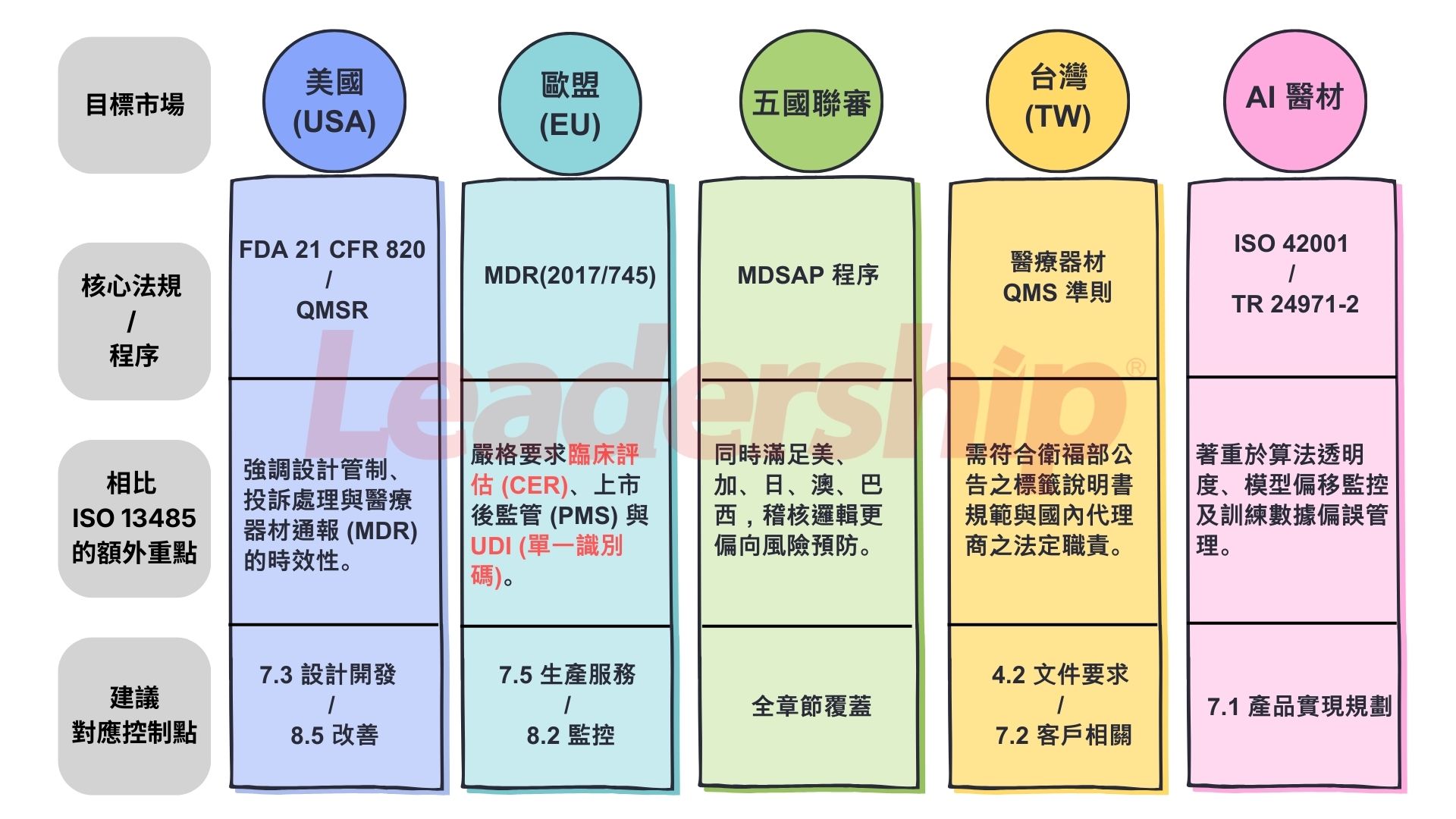
- 美國 FDA: 提出 QMSR 修正案,將品質系統全面與 ISO 13485 接軌,但強化了設計管制與通報時效。
- 歐盟: 透過 EN ISO 13485 調和版應對嚴苛的 MDR 要求,重點在於上市後監管 (PMS) 與臨床評價。
- MDSAP: 涵蓋五國審核程序的單一審核程序。
- 台灣: 現行的「醫療器材品質管理系統準則(QMS)」亦以此為根基。
四大科技浪潮:傳統品管系統的「水土不服」
標準雖然不變,但我們生產的產品早已脫胎換骨。如果您的 QMS 還停留在「實體製造」思維,將難以應對以下四大衝突:
- 人工智慧(AI): 傳統 QMS 習慣「凍結設定」後上市,但 AI 演算法會持續學習。您需要建立對應的變更管理(Change Control),否則在稽核時極易被判定為驗證失效。
- 醫材軟體(SaMD): 當軟體就是產品本身,傳統「進料、組裝、檢驗」流程已不再適用。若未銜接 IEC 62304 規範,品質系統將出現巨大的管理斷層。
- 互聯網與資安防禦: 資安不再只是 IT 部門的事。FDA 現在要求醫材必須具備 SBOM(軟體材料清單),若供應商管理未納入此項,將面臨產品無法通關的風險。
- 3D 列印: 從「批量生產」轉向「客製化」,您的「批次追溯」與「製程驗證」定義必須重新定義。
行動對策:活用 TR 與 TS 補足規範缺口
既然 ISO 13485 本體不動,聰明的業者應該轉向技術報告(TR)與技術規範(TS)。這些指南才是目前補齊現行規範缺口的關鍵拼圖:
關鍵指南工具箱: * 參考 ISO/TS 23485 了解標準本體的最新應用。
- 針對 AI 機器學習,應導入 ISO/TS 24971-2 進行風險管理。
- 針對 可用性工程,應遵循 IEC/CD TS 62366-2 以確保數位化操作的安全性。
數位化時代的品管落實建議:
- 雲端資料與資安控管: 透過雲端系統協助多據點資料交接,但須嚴格執行存取控制並遵循 ALCOA+ 原則,確保品質數據的即時性、完整性與不可竄改性。
- AI 品質與風險監控: 建立 AI 管理機制,利用 ISO 42001 評估 AI 有效性。重點在於監測「模型漂移(Model Drift)」與效能退化,確保模型更新時依循既定的風險控制流程。
- 強化上市後監控 (PMS): 依據 ISO/AWI TR 20416 持續收集表現數據。針對 AI 模型,需在部署後監視其準確度與偏倚,及早發現新出現的偏差。
專業工具箱:ISO 13485 核心架構下的「國別外掛」清單
結語:條文是死的,管理是活的
ISO 13485 預計近五年內不會改版,但這並不代表您可以停下腳步。
關鍵不在於條文何時更新,而在於您如何主動將 AI、資安、數據完整性等新興風險,納入既有的 QMS 架構中。在這個法規趨嚴、技術競爭加劇的環境下,唯有「超前部署」國際指南(TR/TS),才能確保您的產品在全球市場中立於不敗之地。
相關文章
前往了解更多我們的輔導項目
![]()
品質管理
![]()
社會責任
![]()
環境保護
![]()
車用標準
![]()
永續ESG
![]()
資訊安全
![]()
食品安全
![]()
供應鏈安全與反恐
![]()
服務驗證與神秘客
![]()
實驗室與技術認證
![]()
企業管理與經營優化